Patrycja Grala, Katarzyna Błońska
Należy zdiagnozować metodami teoretycznymi pewne
zjawisko i postawić hipotezę popartą naukowymi dowodami -
Analiza bioinformatyczna.
Po ciężkim ataku grypy po kilku
godzinach/dniach poprawy nagle niektórzy pacjenci zapadają na inną chorobę,
która bardzo często prowadi do śmierci. Objawami choroby są: ogólna apatia,
słabość, utraty przytmności, śpiączki, bardzo silne blednięcie, niedokrwistość.
Śmierć następuje w skutek osłabienia organizmu, niedokrwistośći, marskości
nażądów wewnętrznych, niewybudzenie ze spiączki, niedotlenienie mózgu. W czasie
sekcji zwłok wykrywano liczne zakrzepy wewnątrz naczyń krwionośnych, w których
stwierdzono silnie zniszczone i zdeformowane erytrocyty oraz bardzo duży udział
immunoglobulin w kompleksach ze specyficznymi peptydami.
W trakcie analiz
immunologicznych pacjentów jeszcze za życia zidentyfikowano peptyd wirusa
(fragment kapsydu), który rozpoznawany jest przez limfocyty T. Do tej pory
odkryto kilka typów tych peptydów i podzielono je ze względu na nasilenie
objawów grypy oraz zakres powikłań pogrypowych. Vir1 - najsilniejsze objawy
grypy, prawie wszyscy pacjenci zmarli w wyniku powikłań, Vir2 - lżejszy przebieg
grypy, prawie wszyscy pacjenci zmarli, Vir3 - lekki przebieg grypy, część
pacjentów (około 60%) zmarło. Stwierdzono również, że w krajach tropikalnych
część pacjentów odpornych na malarię jest również odporna na powikłania
pochorobowe, podobną odporność ma niewielka część ludzi z innych klimatów
cierpiących na niedokrwistość wrodzoną.
Sekwencje kapsydów
>vir_1A
VHLTPEEKSAVTALWGKVNVDEVGGEALGR
>vir_1B
LTPEEKSAVTALWGKVNVDEVGGEALGR
>vir_2A
LTPEESGVTALWGKVNVDEVEALGR
>vir_2B
HITPEESGVTALWGKVNVDEVEALGR
>vir_2C
TPEEKSGVTALWGKVNVDEVEALGR
>vir_3A
AITPEEKSAGAVTAIWAKVNVDE
>vir_3B
ALHPEEKSAGAAVTAIWANVDEV
>vir_3C
MITPEEKSAGAVTAIWGVDE
Wyniki analizy ClustalW i BLASTP 2.2.24+
Wyniki analizy ClustalW
SeqA Name Len(aa) SeqB Name Len(aa) Score
===================================================
1 vir_1A 30 2 vir_1B 28 100
1 vir_1A 30 3 vir_2A 25 60
1 vir_1A 30 4 vir_2B 26 57
1 vir_1A 30 5 vir_2C 25 76
1 vir_1A 30 6 vir_3A 23 73
1 vir_1A 30 7 vir_3B 23 52
1 vir_1A 30 8 vir_3C 20 60
2 vir_1B 28 3 vir_2A 25 60
2 vir_1B 28 4 vir_2B 26 57
2 vir_1B 28 5 vir_2C 25 76
2 vir_1B 28 6 vir_3A 23 73
2 vir_1B 28 7 vir_3B 23 52
2 vir_1B 28 8 vir_3C 20 60
3 vir_2A 25 4 vir_2B 26 96
3 vir_2A 25 5 vir_2C 25 84
3 vir_2A 25 6 vir_3A 23 43
3 vir_2A 25 7 vir_3B 23 21
3 vir_2A 25 8 vir_3C 20 25
4 vir_2B 26 5 vir_2C 25 84
4 vir_2B 26 6 vir_3A 23 43
4 vir_2B 26 7 vir_3B 23 21
4 vir_2B 26 8 vir_3C 20 25
5 vir_2C 25 6 vir_3A 23 43
5 vir_2C 25 7 vir_3B 23 21
5 vir_2C 25 8 vir_3C 20 25
6 vir_3A 23 7 vir_3B 23 69
6 vir_3A 23 8 vir_3C 20 75
7 vir_3B 23 8 vir_3C 20 65
===================================================

WNIOSKI
Można stwierdzić że sekwencje powtarzające się (konseratywne) w każdym typie wirusa kodują białko rozpoznawane
przez ludzki układ odpornościowy a sekwencja EALGR odpowiada za zwiększoną letalność u wirusów typu 1 i 2.
Sekwencja aminokwasowa EALGR znajdujšca się w Vir1 i Vir2 może świadczyć o ich większej zjadliwości, co może doprowadzić do śmierci pacjentów,
zaś brak tej sekwencji u Vir3 może objawić się mniejszš śmiertelnościš.
W podanych wyżej sekwencjach aminokwasowych możemy zauważyć zgodność 3 bloków aminokwasowych u wszystkich typów
wirusów, co najprawdopodobniej świadczy o zdolności ich łšczenia się z immunoglobulinami.
Wyniki BLASTP 2.2.24+ :
Znalezione sekwencje które pokrywały się z sekwencjami wirusa:
Chain B, Human Hemoglobin A Mutant Beta H63w Carbonmonoxy-F
beta globin [Homo sapiens]
Chain D, Neutron Structure Analysis Of Deoxy Human Hemoglobin
truncated beta globin [Homo sapiens] >gb|ACF16772.1| truncated b
mutant beta-globin [Homo sapiens]
WNIOSKI PO DOKONANEJ ANALIZIE SEKWENCJI
Dzięki wykonanym analizom możemy wywnioskować, że sekwencje wirusów sa homologoczne do cząsteczki hemoglobiny.
Podobieństwo tych sekwencji może świadczyć o wspólnym pochodzeniu. Zatem, gdy w organizmie dojdzie do zakażenia,
którymkolwiek z wyżej wymienionych wirusów, wówczas nasz system odpornościowy może (oprócz obcych czšsteczek)
rozpoznawać jako obcš właśnie hemoglobinę, co może następnie doprowadzić do chorób autoimmunologicznych.
W takiej sytuacji, przeciwciała łączą się również z hemoglobiną i mogą naznaczyć je w celu zniszczenia.
Jedynie u osób chorych na anemię sierpowatš dzięki temu, że majš zmienionš sekwencję aminokwasowš hemoglobiny.
przeciwciała nie łączą się z nią (hemoglobiną), w wyniku czego nie dochodzi do agresji przeciw własnemu organizmowi
Poza tym Wirulentność najprawdopodobniej zależy od sekwencji aminokwasowej białek kapsydu. W sekwencji tej możemy
zaobserwować regiony konserwatywne obecne u wszystkich analizowanych wirusów. Są to sekwencje, które odpowiadają
przede wszystkim za immunogenność. Poza tym, wszystkie analizowane sekwencje aminokwasowe wykazujš podobieństwo
do sekwencji łańcuchów ludzkiej beta hemoglobiny. Po zwalczaniu wirusa przeciwciała mogą być skierowane przeciw
ludzkim białkom o sekwencji homologicznej do sekwencji białek wirusowych, a to może wywołać chorobę o podłożu
autoimmunoagresji.
Analiza przy pomocy programu RasMol:
czasteczka hemoglobiny

hemoglobina z zaznaczonym lancuchami B,
NA PODSTAWIE MULTIPLE ALIGNMENT, W CZĽSTECZCE HEMOGLOBINY NA ŁAŃCUCHU BETA ZAZNACZONO SEKWENCJE KONSERWATYWNE WZGLĘDEM SEKWENCJI WIRUSÓW
Sekwencja homologiczna dla hemoglobiny i wir1
Sekwencja homologiczna dla hemoglobiny i wir2
Sekwencja homologiczna dla hemoglobiny i wir3
Hydrofobowość
Na podstawie hydrofobowości aminokwasów według Kyte&Doolittle sporzadzono profil hydrofobowosci hemoglobiny
Struktura hemoglobiny (1HBA).Gradient kolorów poszczególnych atomów odpowiada dokładnie wartości hydrofobowości
aminokwasów:(najciemniejszy niebieski- aminokwasy najbardziej hydrofilne (ciemne - wartości najwyższe, jasne -
wartości najniższe hydrofobowości/hydrofilowości). Woda zaznaczona jako gwiazdki
; najciemniejszy czerwony-
aminokwasy najbardziej hydrofobowe)

Skrypt
Do stworzenia profilu hydrofobowości tej molekuły w programie RASMOL wykorzystano sporzšdzony w notatniku skrypt
o następujšcych komendach:
select arg
color [000,000,250]
select lys
color [000,000,221]
select asp
color [000,000,199]
select glu
color [000,000,199]
select asn
color [000,000,199]
select gln
color [000,000,199]
select his
color [000,000,175]
select pro
color [000,000,079]
select tyr
color [000,000,063]
select try
color [000,000,045]
select scr
color [000,000,039]
select thr
color [000,000,033]
select gly
color [000,000,009]
select ala
color [250,000,000]
select cys
color [238,000,000]
select phe
color [215,000,000]
select ile
color [159,000,000]
select leu
color [142,000,000]
select met
color [108,000,000]
select val
color [102,000,000]
select all
spacefill
select water
color yellow
stars
Tabela ExPASy tools
Dzięki ExPASy tools możliwe było wykonanie szeregu czynności prowadzšcych do poznania między innymi
budowy, cech, a także właściwości analizowanych molekuł (sekwencji).
| Nazwa | Sekwencja aa | Sekwencja DNA | Ip | molWt | Aa composit. | Hydrofob + profil | Hydrofil + profil | Antygen + profil | Second. Struct. |
| 1HBB | VHLTPEEKSAVTALWGKVNVDEVGGEALGR | gtgcatctgaccccggaagaaaaaagcgcggtgaccgcgctgtggggcaaagtgaacgtg gatgaagtgggcggcgaagcgctgggccgc | 4.96 | 3162.55 | Ala (A) 3 10.0%
Arg (R) 1 3.3%
Asn (N) 1 3.3%
Asp (D) 1 3.3%
Cys (C) 0 0.0%
Gln (Q) 0 0.0%
Glu (E) 4 13.3%
Gly (G) 4 13.3%
His (H) 1 3.3%
Ile (I) 0 0.0%
Leu (L) 3 10.0%
Lys (K) 2 6.7%
Met (M) 0 0.0%
Phe (F) 0 0.0%
Pro (P) 1 3.3%
Ser (S) 1 3.3%
Thr (T) 2 6.7%
Trp (W) 1 3.3%
Tyr (Y) 0 0.0%
Val (V) 5 16.7%
Pyl (O) 0 0.0%
Sec (U) 0 0.0%
(B) 0 0.0%
(Z) 0 0.0%
(X) 0 0.0% | MIN: -1.289 MAX: 1.100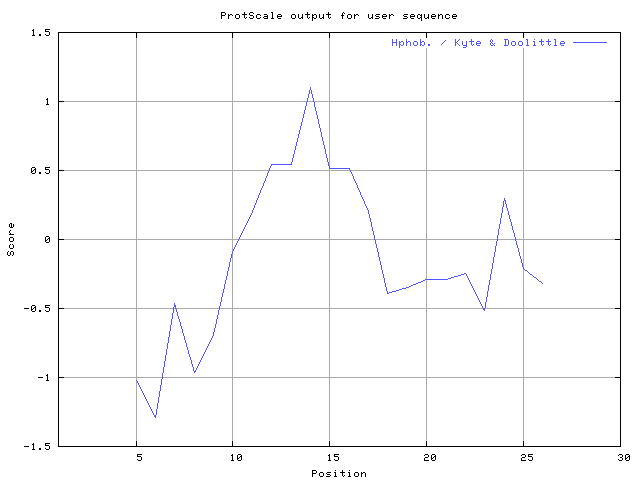 | MIN: -0.733 MAX: 0.722  | Max_score_pos at "*"
(1) Score 1.087 length 16 at residues 8->23
*
Sequence: KSAVTALWGKVNVDEV
| |
8 23
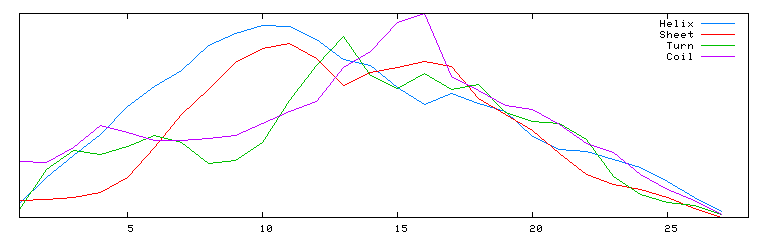
| SOPMA :
Alpha helix (Hh) : 12 is 40.00%
310 helix (Gg) : 0 is 0.00%
Pi helix (Ii) : 0 is 0.00%
Beta bridge (Bb) : 0 is 0.00%
Extended strand (Ee) : 3 is 10.00%
Beta turn (Tt) : 4 is 13.33%
Bend region (Ss) : 0 is 0.00%
Random coil (Cc) : 11 is 36.67%
Ambigous states (?) : 0 is 0.00%
Other states : 0 is 0.00%
 |
| Vir_1A | VHLTPEEKSAVTALWGKVNVDEVGGEALGR | gtgcatctgaccccggaagaaaaaagcgcggtgaccgcgctgtggggcaaagtgaacgtg gatgaagtgggcggcgaagcgctgggccgc | 4.96 | 3162.55 | Ala (A) 3 10.0%
Arg (R) 1 3.3%
Asn (N) 1 3.3%
Asp (D) 1 3.3%
Cys (C) 0 0.0%
Gln (Q) 0 0.0%
Glu (E) 4 13.3%
Gly (G) 4 13.3%
His (H) 1 3.3%
Ile (I) 0 0.0%
Leu (L) 3 10.0%
Lys (K) 2 6.7%
Met (M) 0 0.0%
Phe (F) 0 0.0%
Pro (P) 1 3.3%
Ser (S) 1 3.3%
Thr (T) 2 6.7%
Trp (W) 1 3.3%
Tyr (Y) 0 0.0%
Val (V) 5 16.7%
Pyl (O) 0 0.0%
Sec (U) 0 0.0%
(B) 0 0.0%
(Z) 0 0.0%
(X) 0 0.0% | MIN: -1.289 MAX: 1.100 | MIN: -0.733 MAX: 0.722 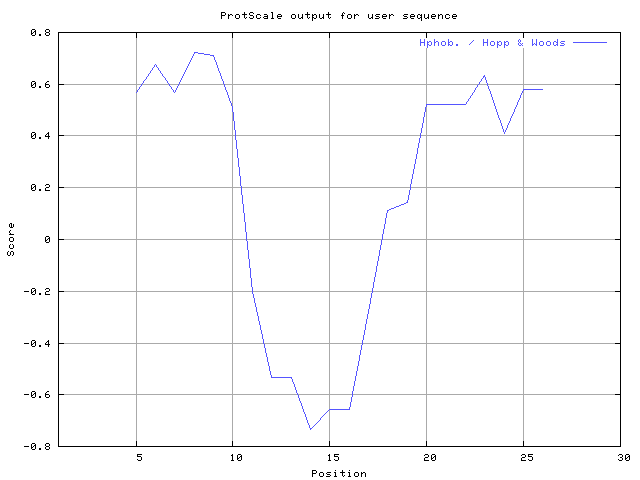 | Max_score_pos at "*"
(1) Score 1.087 length 16 at residues 8->23
*
Sequence: KSAVTALWGKVNVDEV
| |
8 23
| SOPMA :
Alpha helix (Hh) : 12 is 40.00%
310 helix (Gg) : 0 is 0.00%
Pi helix (Ii) : 0 is 0.00%
Beta bridge (Bb) : 0 is 0.00%
Extended strand (Ee) : 3 is 10.00%
Beta turn (Tt) : 4 is 13.33%
Bend region (Ss) : 0 is 0.00%
Random coil (Cc) : 11 is 36.67%
Ambigous states (?) : 0 is 0.00%
Other states : 0 is 0.00%
 |
| Vir_1B | LTPEEKSAVTALWGKVNVDEVGGEALGR | ctgaccccggaagaaaaaagcgcggtgaccgcgctgtggggcaaagtgaacgtggatgaa gtgggcggcgaagcgctgggccgc | 4.59 | 2926.28 | Ala (A) 3 10.7%
Arg (R) 1 3.6%
Asn (N) 1 3.6%
Asp (D) 1 3.6%
Cys (C) 0 0.0%
Gln (Q) 0 0.0%
Glu (E) 4 14.3%
Gly (G) 4 14.3%
His (H) 0 0.0%
Ile (I) 0 0.0%
Leu (L) 3 10.7%
Lys (K) 2 7.1%
Met (M) 0 0.0%
Phe (F) 0 0.0%
Pro (P) 1 3.6%
Ser (S) 1 3.6%
Thr (T) 2 7.1%
Trp (W) 1 3.6%
Tyr (Y) 0 0.0%
Val (V) 4 14.3%
Pyl (O) 0 0.0%
Sec (U) 0 0.0%
(B) 0 0.0%
(Z) 0 0.0%
(X) 0 0.0% | MIN: -0.967 MAX: 1.100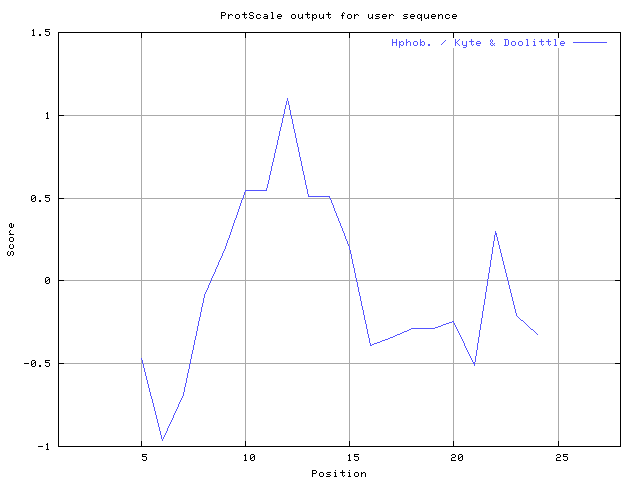 | MIN: -0.733 MAX: 0.722 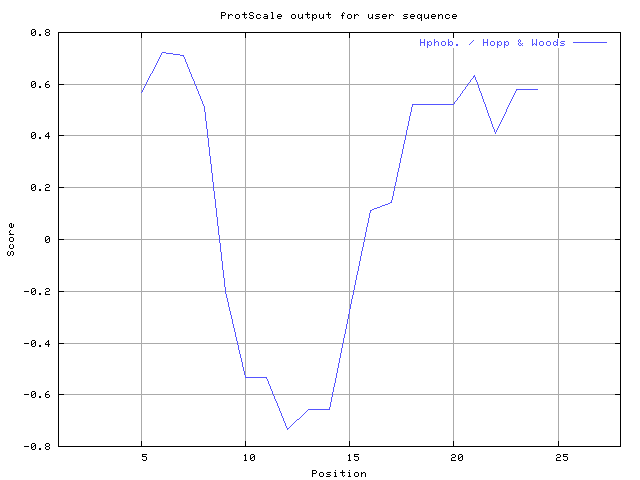 | Max_score_pos at "*"
(1) Score 1.087 length 16 at residues 6->21
*
Sequence: KSAVTALWGKVNVDEV
| |
6 21 | SOPMA :
Alpha helix (Hh) : 12 is 42.86%
310 helix (Gg) : 0 is 0.00%
Pi helix (Ii) : 0 is 0.00%
Beta bridge (Bb) : 0 is 0.00%
Extended strand (Ee) : 1 is 3.57%
Beta turn (Tt) : 4 is 14.29%
Bend region (Ss) : 0 is 0.00%
Random coil (Cc) : 11 is 39.29%
Ambigous states (?) : 0 is 0.00%
Other states : 0 is 0.00%
 |
| Vir_2A | LTPEESGVTALWGKVNVDEVEALGR | ctgaccccggaagaaagcggcgtgaccgcgctgtggggcaaagtgaacgtggatgaagtg gaagcgctgggccgc | 4.25 | 2669.97 | Ala (A) 2 8.0%
Arg (R) 1 4.0%
Asn (N) 1 4.0%
Asp (D) 1 4.0%
Cys (C) 0 0.0%
Gln (Q) 0 0.0%
Glu (E) 4 16.0%
Gly (G) 3 12.0%
His (H) 0 0.0%
Ile (I) 0 0.0%
Leu (L) 3 12.0%
Lys (K) 1 4.0%
Met (M) 0 0.0%
Phe (F) 0 0.0%
Pro (P) 1 4.0%
Ser (S) 1 4.0%
Thr (T) 2 8.0%
Trp (W) 1 4.0%
Tyr (Y) 0 0.0%
Val (V) 4 16.0%
Pyl (O) 0 0.0%
Sec (U) 0 0.0%
(B) 0 0.0%
(Z) 0 0.0%
(X) 0 0.0% | MIN: -0.633 MAX: 0.856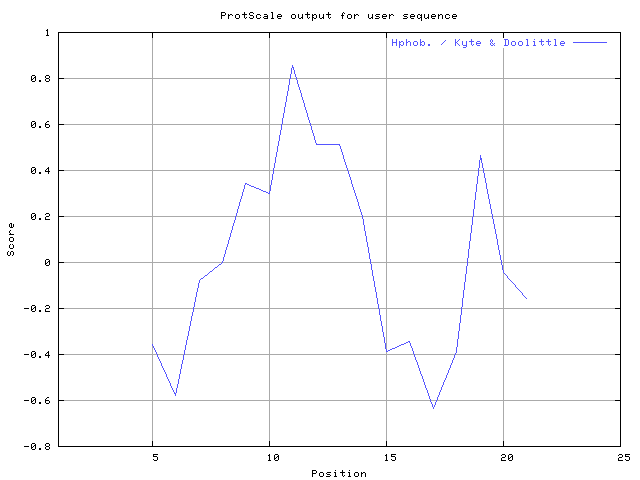 | MIN: -0.678 MAX: 0.856 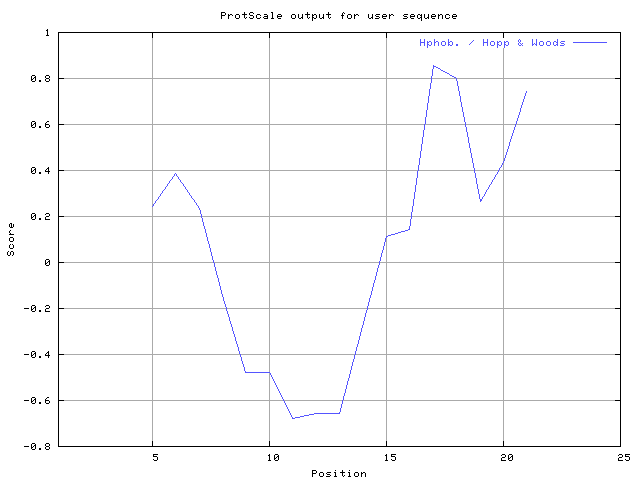 |
Max_score_pos at "*"
(1) Score 1.093 length 15 at residues 8->22
*
Sequence: VTALWGKVNVDEVEA
| |
8 22 | SOPMA :
Alpha helix (Hh) : 14 is 56.00%
310 helix (Gg) : 0 is 0.00%
Pi helix (Ii) : 0 is 0.00%
Beta bridge (Bb) : 0 is 0.00%
Extended strand (Ee) : 1 is 4.00%
Beta turn (Tt) : 2 is 8.00%
Bend region (Ss) : 0 is 0.00%
Random coil (Cc) : 8 is 32.00%
Ambigous states (?) : 0 is 0.00%
Other states : 0 is 0.00%
 |
| Vir_2B | HITPEESGVTALWGKVNVDEVEALGR | catattaccccggaagaaagcggcgtgaccgcgctgtggggcaaagtgaacgtggatgaa gtggaagcgctgggccgc | 4.57 | 2807.11 | Ala (A) 2 7.7%
Arg (R) 1 3.8%
Asn (N) 1 3.8%
Asp (D) 1 3.8%
Cys (C) 0 0.0%
Gln (Q) 0 0.0%
Glu (E) 4 15.4%
Gly (G) 3 11.5%
His (H) 1 3.8%
Ile (I) 1 3.8%
Leu (L) 2 7.7%
Lys (K) 1 3.8%
Met (M) 0 0.0%
Phe (F) 0 0.0%
Pro (P) 1 3.8%
Ser (S) 1 3.8%
Thr (T) 2 7.7%
Trp (W) 1 3.8%
Tyr (Y) 0 0.0%
Val (V) 4 15.4%
Pyl (O) 0 0.0%
Sec (U) 0 0.0%
(B) 0 0.0%
(Z) 0 0.0%
(X) 0 0.0% | MIN: -0.633 MAX: 0.856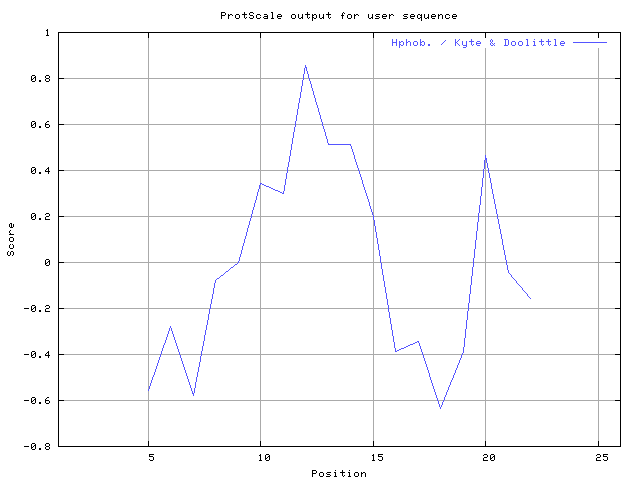 | MIN: -0.678 MAX: 0.856 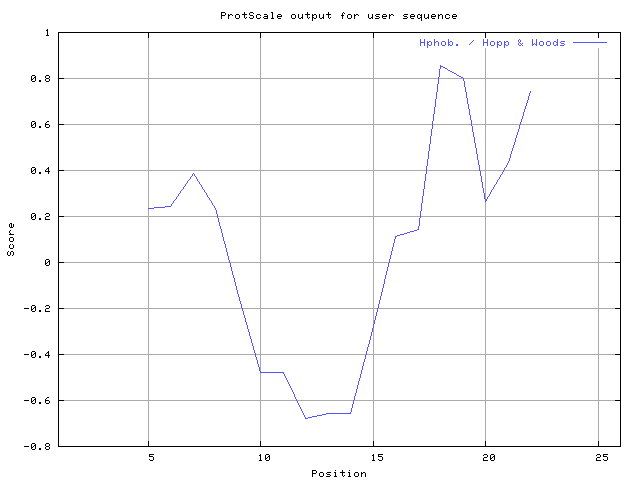 | Max_score_pos at "*"
(1) Score 1.093 length 15 at residues 9->23
*
Sequence: VTALWGKVNVDEVEA
| |
9 23 | SOPMA :
Alpha helix (Hh) : 14 is 53.85%
310 helix (Gg) : 0 is 0.00%
Pi helix (Ii) : 0 is 0.00%
Beta bridge (Bb) : 0 is 0.00%
Extended strand (Ee) : 2 is 7.69%
Beta turn (Tt) : 2 is 7.69%
Bend region (Ss) : 0 is 0.00%
Random coil (Cc) : 8 is 30.77%
Ambigous states (?) : 0 is 0.00%
Other states : 0 is 0.00%
 |
| Vir_2C | TPEEKSGVTALWGKVNVDEVEALGR | accccggaagaaaaaagcggcgtgaccgcgctgtggggcaaagtgaacgtggatgaagtg gaagcgctgggccgc | 4.58 | 2684.99 | Ala (A) 2 8.0%
Arg (R) 1 4.0%
Asn (N) 1 4.0%
Asp (D) 1 4.0%
Cys (C) 0 0.0%
Gln (Q) 0 0.0%
Glu (E) 4 16.0%
Gly (G) 3 12.0%
His (H) 0 0.0%
Ile (I) 0 0.0%
Leu (L) 2 8.0%
Lys (K) 2 8.0%
Met (M) 0 0.0%
Phe (F) 0 0.0%
Pro (P) 1 4.0%
Ser (S) 1 4.0%
Thr (T) 2 8.0%
Trp (W) 1 4.0%
Tyr (Y) 0 0.0%
Val (V) 4 16.0%
Pyl (O) 0 0.0%
Sec (U) 0 0.0%
(B) 0 0.0%
(Z) 0 0.0%
(X) 0 0.0%
| MIN: -1.211 MAX: 0.856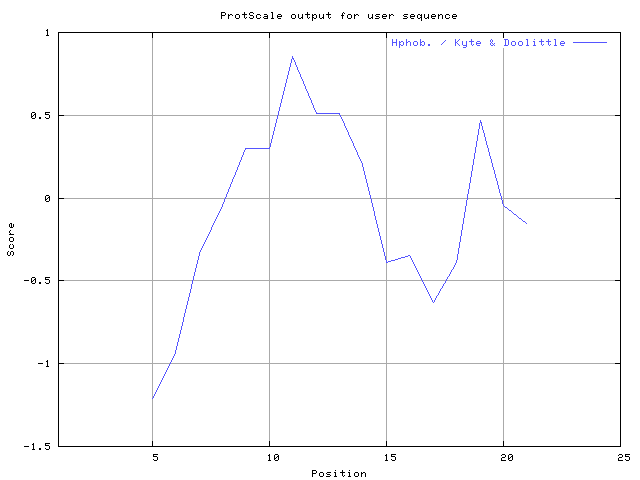 | MIN: -0.678 MAX: 0.856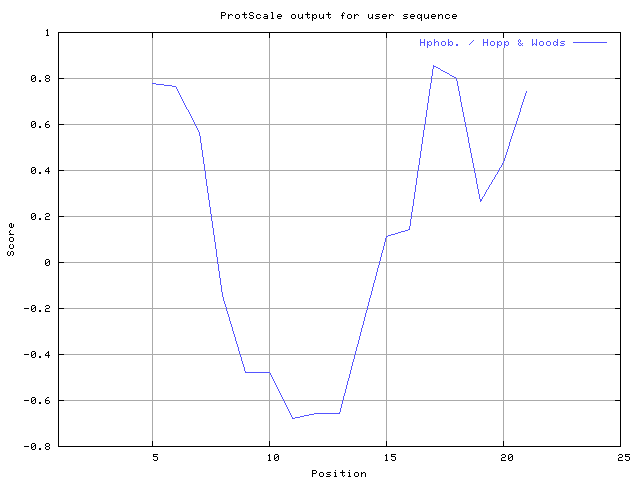 | Max_score_pos at "*"
(1) Score 1.093 length 16 at residues 7->22
*
Sequence: GVTALWGKVNVDEVEA
| |
7 22 | SOPMA :
Alpha helix (Hh) : 15 is 60.00%
310 helix (Gg) : 0 is 0.00%
Pi helix (Ii) : 0 is 0.00%
Beta bridge (Bb) : 0 is 0.00%
Extended strand (Ee) : 1 is 4.00%
Beta turn (Tt) : 2 is 8.00%
Bend region (Ss) : 0 is 0.00%
Random coil (Cc) : 7 is 28.00%
Ambigous states (?) : 0 is 0.00%
Other states : 0 is 0.00%
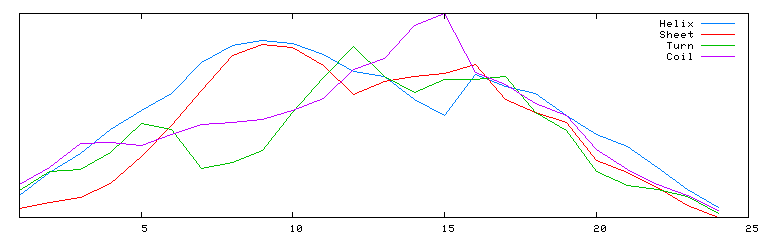 |
| Vir_3A | AITPEEKSAGAVTAIWAKVNVDE | gcgattaccccggaagaaaaaagcgcgggcgcggtgaccgcgatttgggcgaaagtgaac gtggatgaa | 4.58 | 2684.99 | Ala (A) 5 21.7%
Arg (R) 0 0.0%
Asn (N) 1 4.3%
Asp (D) 1 4.3%
Cys (C) 0 0.0%
Gln (Q) 0 0.0%
Glu (E) 3 13.0%
Gly (G) 1 4.3%
His (H) 0 0.0%
Ile (I) 2 8.7%
Leu (L) 0 0.0%
Lys (K) 2 8.7%
Met (M) 0 0.0%
Phe (F) 0 0.0%
Pro (P) 1 4.3%
Ser (S) 1 4.3%
Thr (T) 2 8.7%
Trp (W) 1 4.3%
Tyr (Y) 0 0.0%
Val (V) 3 13.0%
Pyl (O) 0 0.0%
Sec (U) 0 0.0%
(B) 0 0.0%
(Z) 0 0.0%
(X) 0 0.0% | MIN: -1.200 MAX: 1.544 | MIN: -1.011 MAX: 0.878  | Max_score_pos at "*"
(1) Score 1.083 length 12 at residues 9->20
*
Sequence: AGAVTAIWAKVN
| |
9 20 | SOPMA :
Alpha helix (Hh) : 10 is 43.48%
310 helix (Gg) : 0 is 0.00%
Pi helix (Ii) : 0 is 0.00%
Beta bridge (Bb) : 0 is 0.00%
Extended strand (Ee) : 0 is 0.00%
Beta turn (Tt) : 1 is 4.35%
Bend region (Ss) : 0 is 0.00%
Random coil (Cc) : 12 is 52.17%
Ambigous states (?) : 0 is 0.00%
Other states : 0 is 0.00%
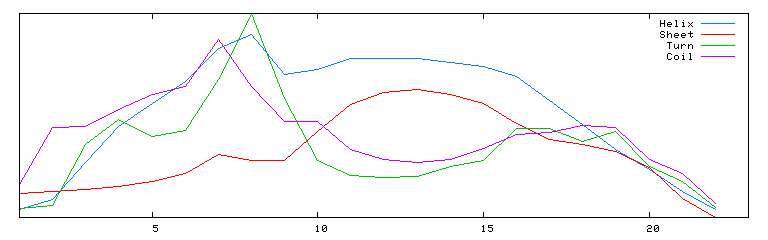 |
| Vir_3B | ALHPEEKSAGAAVTAIWANVDEV | gcgctgcatccggaagaaaaaagcgcgggcgcggcggtgaccgcgatttgggcgaacgtg gatgaagtg | 4.40 | 2378.62 | Ala (A) 6 26.1%
Arg (R) 0 0.0%
Asn (N) 1 4.3%
Asp (D) 1 4.3%
Cys (C) 0 0.0%
Gln (Q) 0 0.0%
Glu (E) 3 13.0%
Gly (G) 1 4.3%
His (H) 1 4.3%
Ile (I) 1 4.3%
Leu (L) 1 4.3%
Lys (K) 1 4.3%
Met (M) 0 0.0%
Phe (F) 0 0.0%
Pro (P) 1 4.3%
Ser (S) 1 4.3%
Thr (T) 1 4.3%
Trp (W) 1 4.3%
Tyr (Y) 0 0.0%
Val (V) 3 13.0%
Pyl (O) 0 0.0%
Sec (U) 0 0.0%
(B) 0 0.0%
(Z) 0 0.0%
(X) 0 0.0% | MIN: -1.478 MAX: 1.556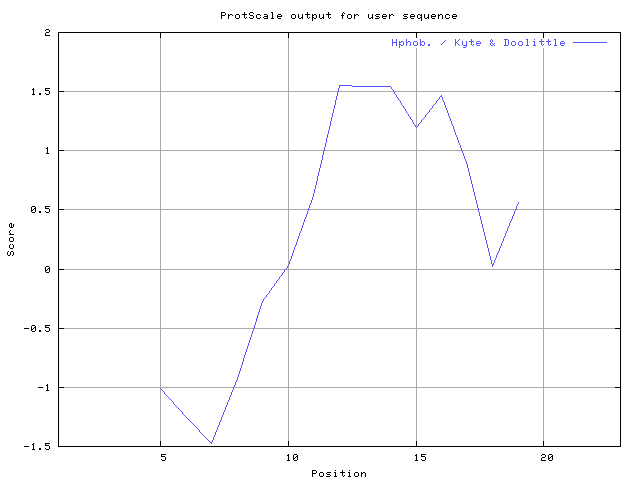 | MIN: -1.100 MAX: 0.867 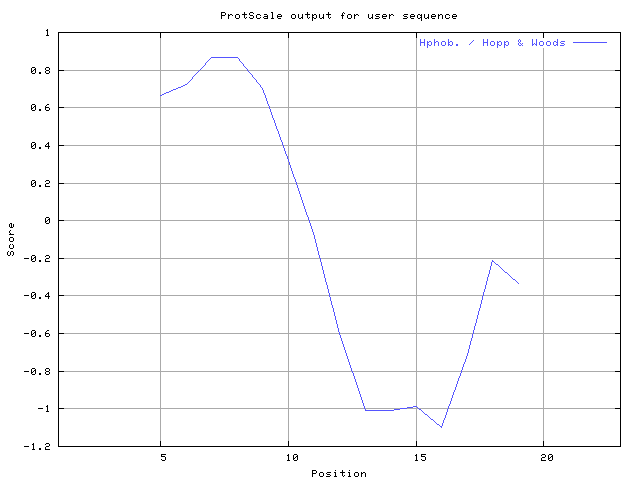 | Max_score_pos at "*"
(1) Score 1.076 length 9 at residues 10->18
*
Sequence: GAAVTAIWA
| |
10 18 | SOPMA :
Alpha helix (Hh) : 17 is 73.91%
310 helix (Gg) : 0 is 0.00%
Pi helix (Ii) : 0 is 0.00%
Beta bridge (Bb) : 0 is 0.00%
Extended strand (Ee) : 0 is 0.00%
Beta turn (Tt) : 0 is 0.00%
Bend region (Ss) : 0 is 0.00%
Random coil (Cc) : 6 is 26.09%
Ambigous states (?) : 0 is 0.00%
Other states : 0 is 0.00%
 |
| Vir_3C | MITPEEKSAGAVTAIWGVDE | atgattaccccggaagaaaaaagcgcgggcgcggtgaccgcgatttggggcgtggatgaa | 4.24 | 2104.36 | Ala (A) 6 26.1%
Arg (R) 0 0.0%
Asn (N) 1 4.3%
Asp (D) 1 4.3%
Cys (C) 0 0.0%
Gln (Q) 0 0.0%
Glu (E) 3 13.0%
Gly (G) 1 4.3%
His (H) 1 4.3%
Ile (I) 1 4.3%
Leu (L) 1 4.3%
Lys (K) 1 4.3%
Met (M) 0 0.0%
Phe (F) 0 0.0%
Pro (P) 1 4.3%
Ser (S) 1 4.3%
Thr (T) 1 4.3%
Trp (W) 1 4.3%
Tyr (Y) 0 0.0%
Val (V) 3 13.0%
Pyl (O) 0 0.0%
Sec (U) 0 0.0%
(B) 0 0.0%
(Z) 0 0.0%
(X) 0 0.0% | MIN: -1.200 MAX: 1.567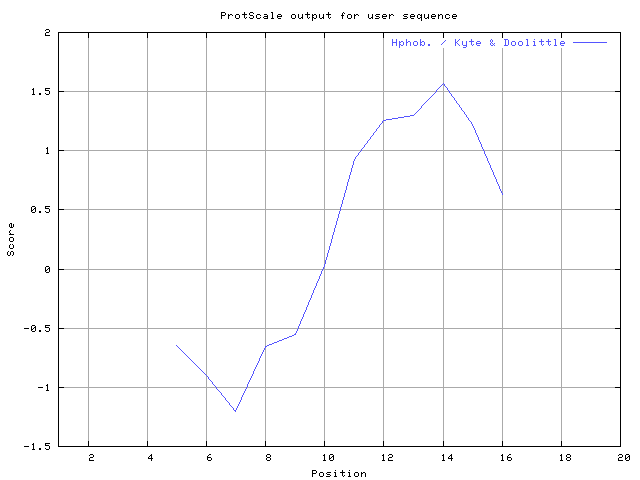 | MIN: -1.067 MAX: 0.878  | Max_score_pos at "*"
(1) Score 1.094 length 9 at residues 9->17
*
Sequence: AGAVTAIWG
| |
9 17 | SOPMA :
Alpha helix (Hh) : 9 is 45.00%
310 helix (Gg) : 0 is 0.00%
Pi helix (Ii) : 0 is 0.00%
Beta bridge (Bb) : 0 is 0.00%
Extended strand (Ee) : 1 is 5.00%
Beta turn (Tt) : 2 is 10.00%
Bend region (Ss) : 0 is 0.00%
Random coil (Cc) : 8 is 40.00%
Ambigous states (?) : 0 is 0.00%
Other states : 0 is 0.00%
 |
Darmowy hosting zapewnia PRV.PL